library(tidyverse)
library(palmerpenguins)
library(marginaleffects)
library(patchwork)
library(parameters)
library(broom)
library(distributional)
library(ggdist)
library(brms)
library(tidybayes)
options(mc.cores = 4) # Use 4 cores for {brms}
theme_set(theme_minimal())
penguins <- penguins |> drop_na(sex)OLS
Typically when we use linear regression (or OLS) to model things in social science, we care about the coefficients so that we can find marginal effects: what happens to the outcome when we move an explanatory variable up and down? Or in the case of categorical explanatory variables, what’s the average outcome across different categories?
Because of this, we don’t typically think about distributions or parameters when working with OLS. BUT WE CAN. And thinking about how outcome distributions change in response to explanatory variables opens up possibilities for modeling all sorts of speical non-normal outcomes.
For instance, we can model the distribution of penguin weights:
ggplot(penguins, aes(x = body_mass_g)) +
geom_density()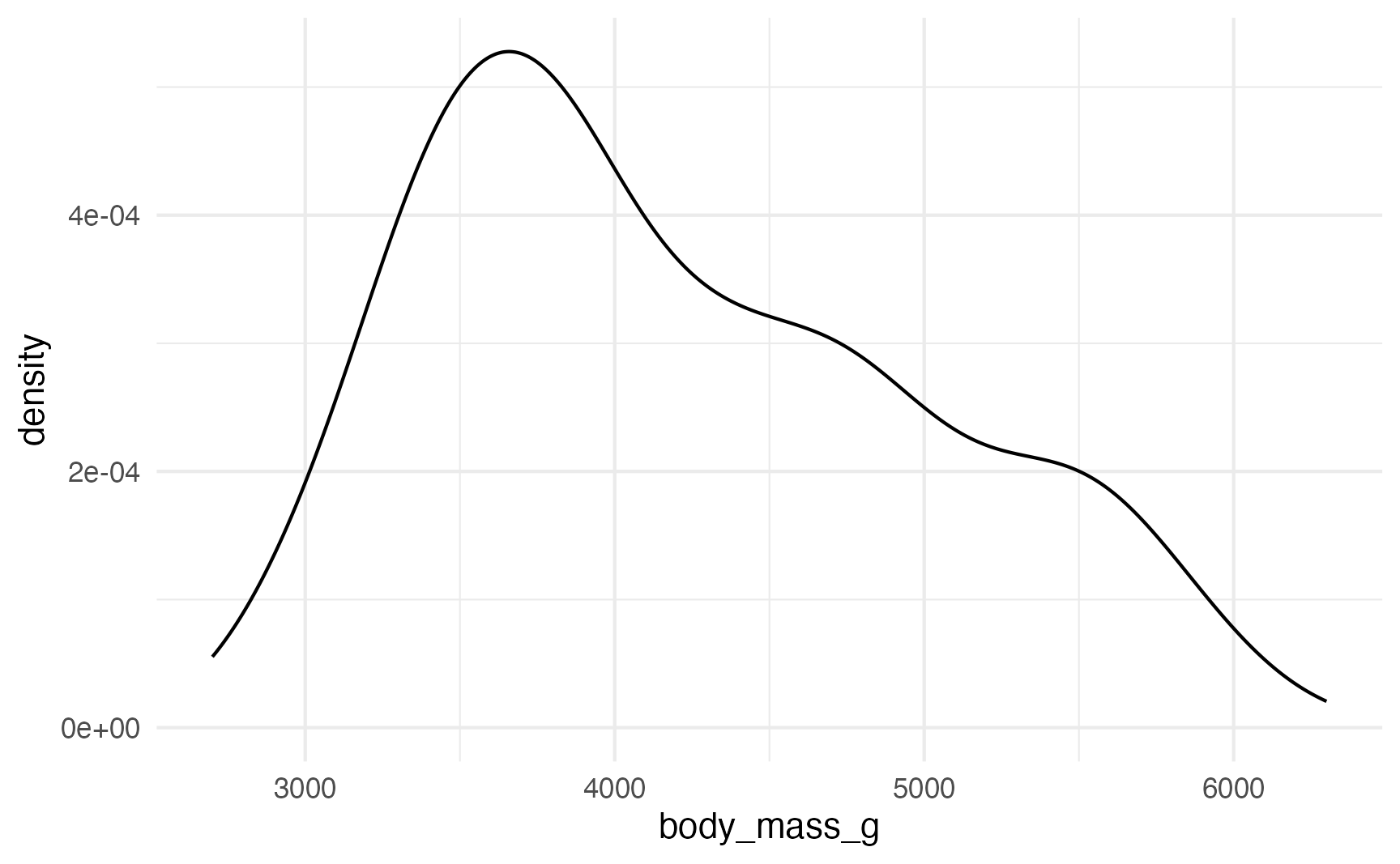
That doesn’t look very normal—it’s skewed right with a long tail. That’s likely because of other factors, like penguin species. Let’s color by species:
ggplot(penguins, aes(x = body_mass_g, color = species)) +
geom_density()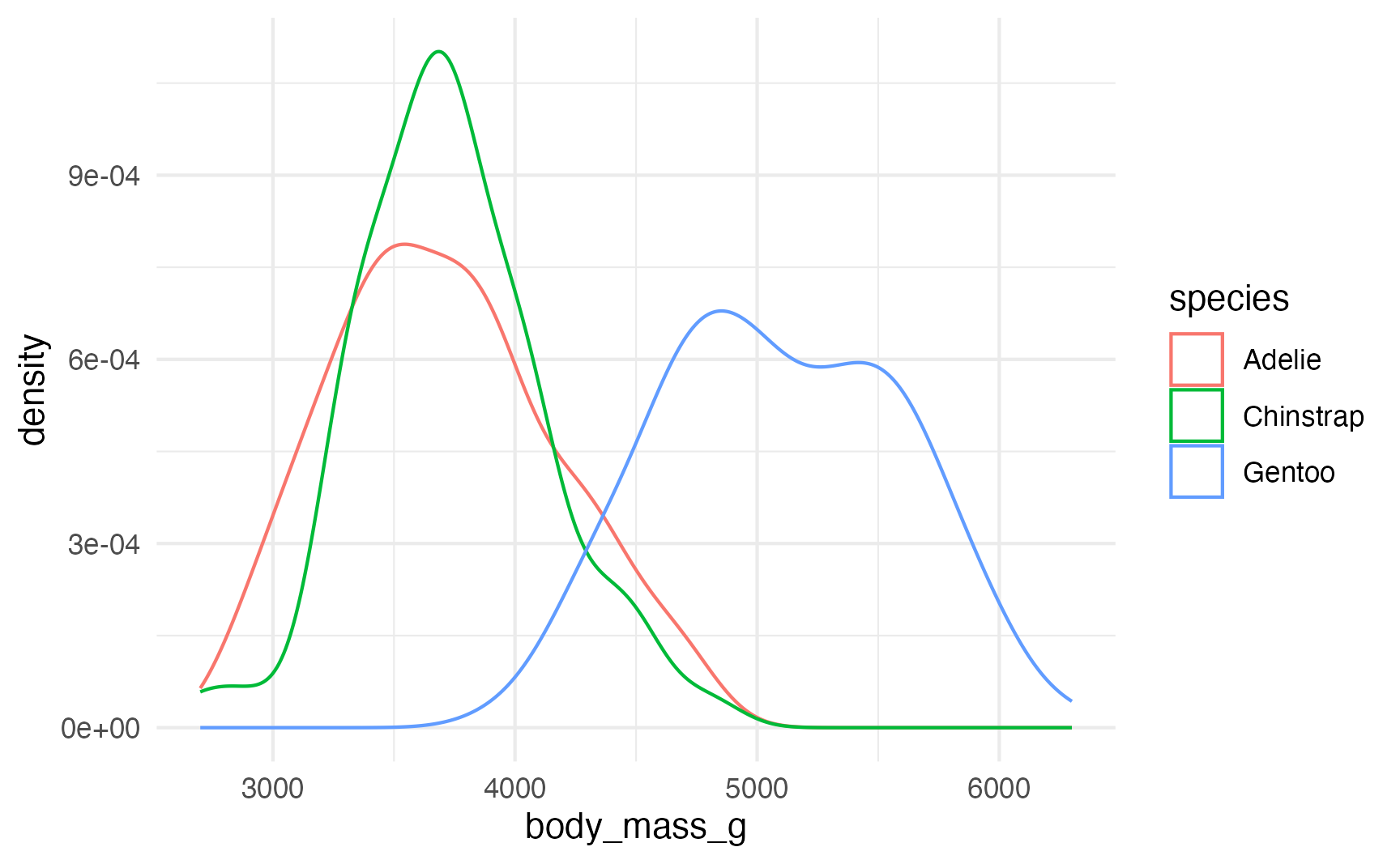
That original lumpy skewed distribution is actually the combination of three separate relatively-normal-looking distributions.
Let’s model body mass based on species:
model <- lm(body_mass_g ~ species, data = penguins)
# parameters::model_parameters() gives a nice summary of the coefficients
model_parameters(model)
## Parameter | Coefficient | SE | 95% CI | t(330) | p
## --------------------------------------------------------------------------------
## (Intercept) | 3706.16 | 38.14 | [3631.14, 3781.18] | 97.18 | < .001
## species [Chinstrap] | 26.92 | 67.65 | [-106.16, 160.01] | 0.40 | 0.691
## species [Gentoo] | 1386.27 | 56.91 | [1274.32, 1498.22] | 24.36 | < .001
##
## Uncertainty intervals (equal-tailed) and p-values (two-tailed) computed
## using a Wald t-distribution approximation.
# So does broom::tidy()
tidy(model, conf.int = TRUE)
## # A tibble: 3 × 7
## term estimate std.error statistic p.value conf.low conf.high
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 (Intercept) 3706. 38.1 97.2 6.88e-245 3631. 3781.
## 2 speciesChinstrap 26.9 67.7 0.398 6.91e- 1 -106. 160.
## 3 speciesGentoo 1386. 56.9 24.4 1.01e- 75 1274. 1498.Since these are all in grams, we can interpret these coefficients directly (and we can think of them as sliders and switches)
It’s a little annoying to have all these estimates be based around Adelie. We can get around that in a couple ways:
Use
marginaleffects::avg_predictions(), which deals with all the relative group averages automatically:avg_predictions(model, variables = "species") ## ## species Estimate Std. Error z Pr(>|z|) S 2.5 % 97.5 % ## Adelie 3706 38.1 97.2 <0.001 Inf 3631 3781 ## Gentoo 5092 42.2 120.6 <0.001 Inf 5010 5175 ## Chinstrap 3733 55.9 66.8 <0.001 Inf 3624 3843 ## ## Type: response ## Columns: species, estimate, std.error, statistic, p.value, s.value, conf.low, conf.highFit a model without the intercept using
0 +:model_sans_intercept <- lm(body_mass_g ~ 0 + species, data = penguins) tidy(model_sans_intercept, conf.int = TRUE) ## # A tibble: 3 × 7 ## term estimate std.error statistic p.value conf.low conf.high ## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> ## 1 speciesAdelie 3706. 38.1 97.2 6.88e-245 3631. 3781. ## 2 speciesChinstrap 3733. 55.9 66.8 8.16e-194 3623. 3843. ## 3 speciesGentoo 5092. 42.2 121. 6.31e-275 5009. 5176.
These coefficients show us the averages for each species, or the \(\mu\) in a normal distribution:
species_averages <- avg_predictions(model, variables = "species")
ggplot(penguins, aes(x = body_mass_g, fill = species)) +
stat_density(geom = "area", alpha = 0.6, position = position_identity()) +
geom_vline(data = species_averages, aes(xintercept = estimate, color = species))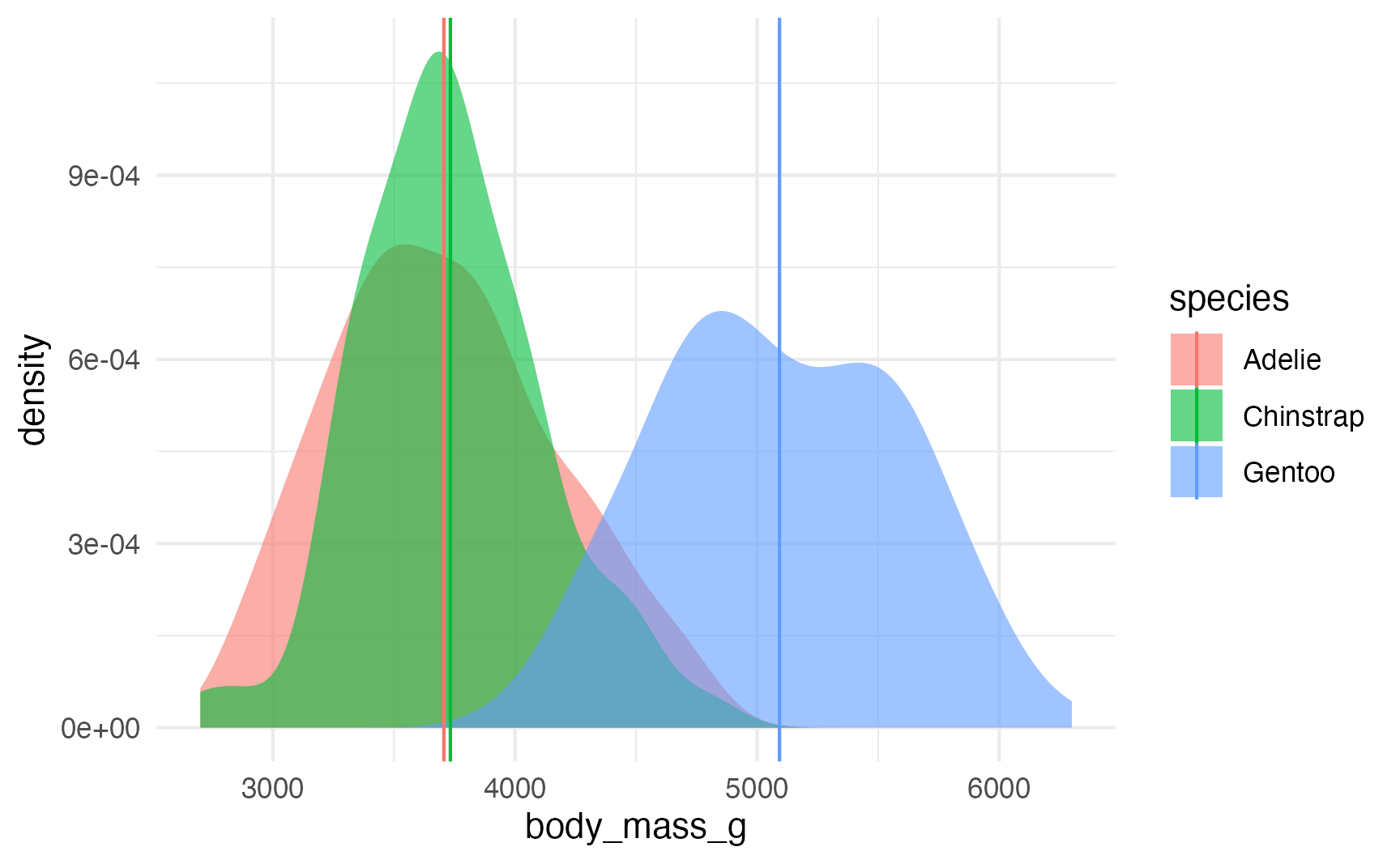
Getting closer to distributional regression
We’re halfway to describing the outcome using distributions—we just need to find the standard deviation for each group.
It’s tempting to use the standard error column in the model results, but that’s wrong! That’s the standard deviation of the coefficient, not the standard deviation for the group.
One way to use the model results to find group-specific distributional standard deviations is to find the standard deviation of the residuals. We can use broom::augment() to add the fitted values and residuals to each row in the original data:
fitted_values <- augment(model)
fitted_values
## # A tibble: 333 × 8
## body_mass_g species .fitted .resid .hat .sigma .cooksd .std.resid
## <int> <fct> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 3750 Adelie 3706. 43.8 0.00685 461. 0.0000209 0.0955
## 2 3800 Adelie 3706. 93.8 0.00685 461. 0.0000960 0.204
## 3 3250 Adelie 3706. -456. 0.00685 461. 0.00227 -0.993
## 4 3450 Adelie 3706. -256. 0.00685 461. 0.000715 -0.558
## 5 3650 Adelie 3706. -56.2 0.00685 461. 0.0000344 -0.122
## 6 3625 Adelie 3706. -81.2 0.00685 461. 0.0000718 -0.177
## 7 4675 Adelie 3706. 969. 0.00685 458. 0.0102 2.11
## 8 3200 Adelie 3706. -506. 0.00685 461. 0.00279 -1.10
## 9 3800 Adelie 3706. 93.8 0.00685 461. 0.0000960 0.204
## 10 4400 Adelie 3706. 694. 0.00685 460. 0.00525 1.51
## # ℹ 323 more rowsWe can then find the standard deviation of the .resid column:
penguin_parameters <- fitted_values |>
group_by(species) |>
summarize(mu = mean(.fitted), sigma = sd(.resid))
penguin_parameters
## # A tibble: 3 × 3
## species mu sigma
## <fct> <dbl> <dbl>
## 1 Adelie 3706. 459.
## 2 Chinstrap 3733. 384.
## 3 Gentoo 5092. 501.These values represent the \(\mu\) and \(\sigma\) for each of the species distributions. We can confirm it with a plot:
ggplot(penguins, aes(x = body_mass_g, fill = species)) +
stat_density(geom = "area", alpha = 0.6, position = position_identity()) +
stat_function(data = penguin_parameters, aes(x = NULL, y = NULL, color = species[1]),
fun = function(x, mu, sigma) dnorm(x, mean = mu, sd = sigma),
args = list(mu = penguin_parameters$mu[1], sigma = penguin_parameters$sigma[1]), size = 1) +
stat_function(data = penguin_parameters, aes(x = NULL, y = NULL, color = species[2]),
fun = function(x, mu, sigma) dnorm(x, mean = mu, sd = sigma),
args = list(mu = penguin_parameters$mu[2], sigma = penguin_parameters$sigma[2]), size = 1) +
stat_function(data = penguin_parameters, aes(x = NULL, y = NULL, color = species[3]),
fun = function(x, mu, sigma) dnorm(x, mean = mu, sd = sigma),
args = list(mu = penguin_parameters$mu[3], sigma = penguin_parameters$sigma[3]), size = 1)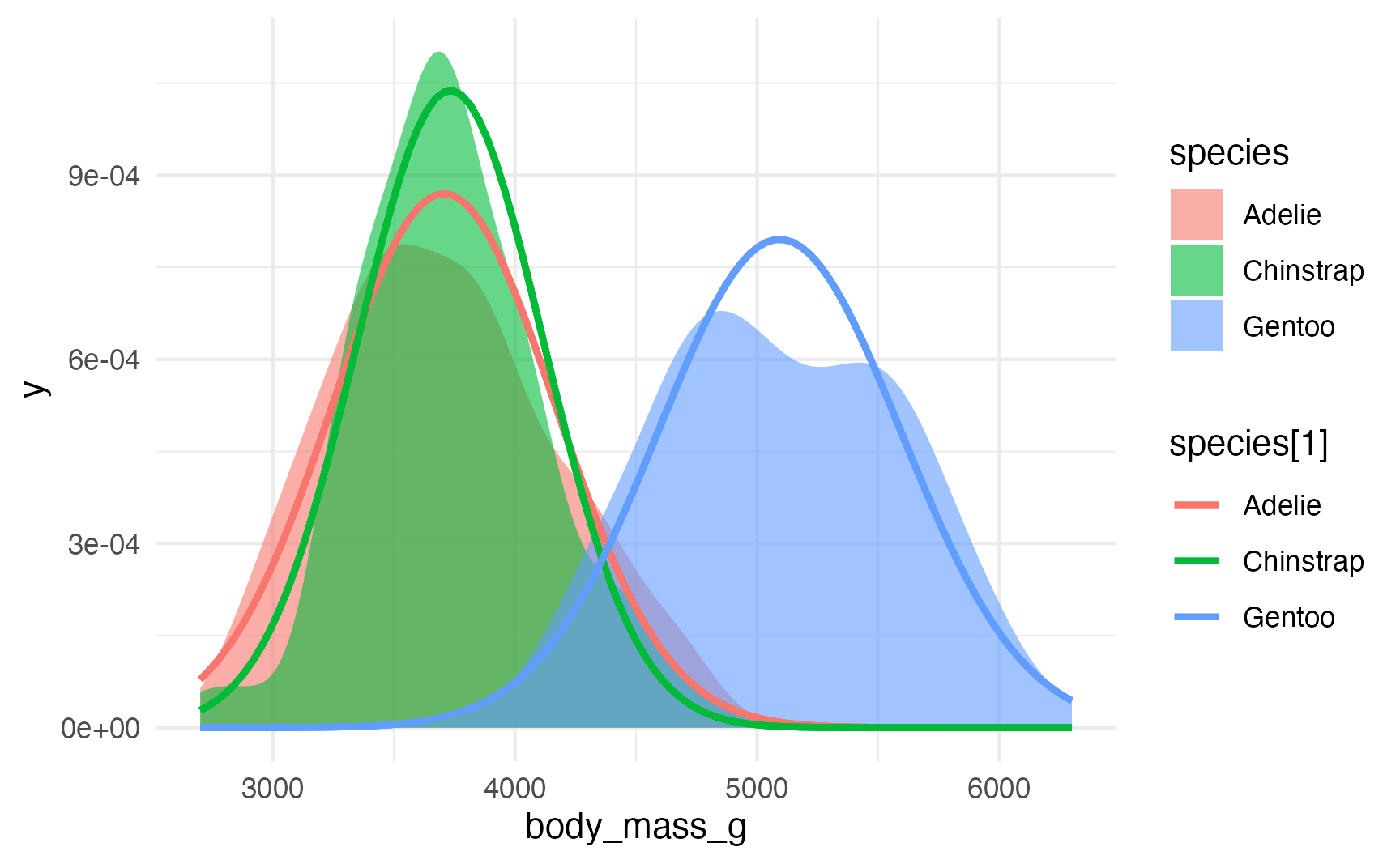
↑ That’s all super gross code though. Instead of adding stat_function() layers manually like that, we can use the {distributional} R package, which lets you work with distribution objects:
penguin_parameters_dist <- penguin_parameters |>
mutate(dist = dist_normal(mu = mu, sigma = sigma))
penguin_parameters_dist
## # A tibble: 3 × 4
## species mu sigma
## <fct> <dbl> <dbl>
## 1 Adelie 3706. 459.
## 2 Chinstrap 3733. 384.
## 3 Gentoo 5092. 501.
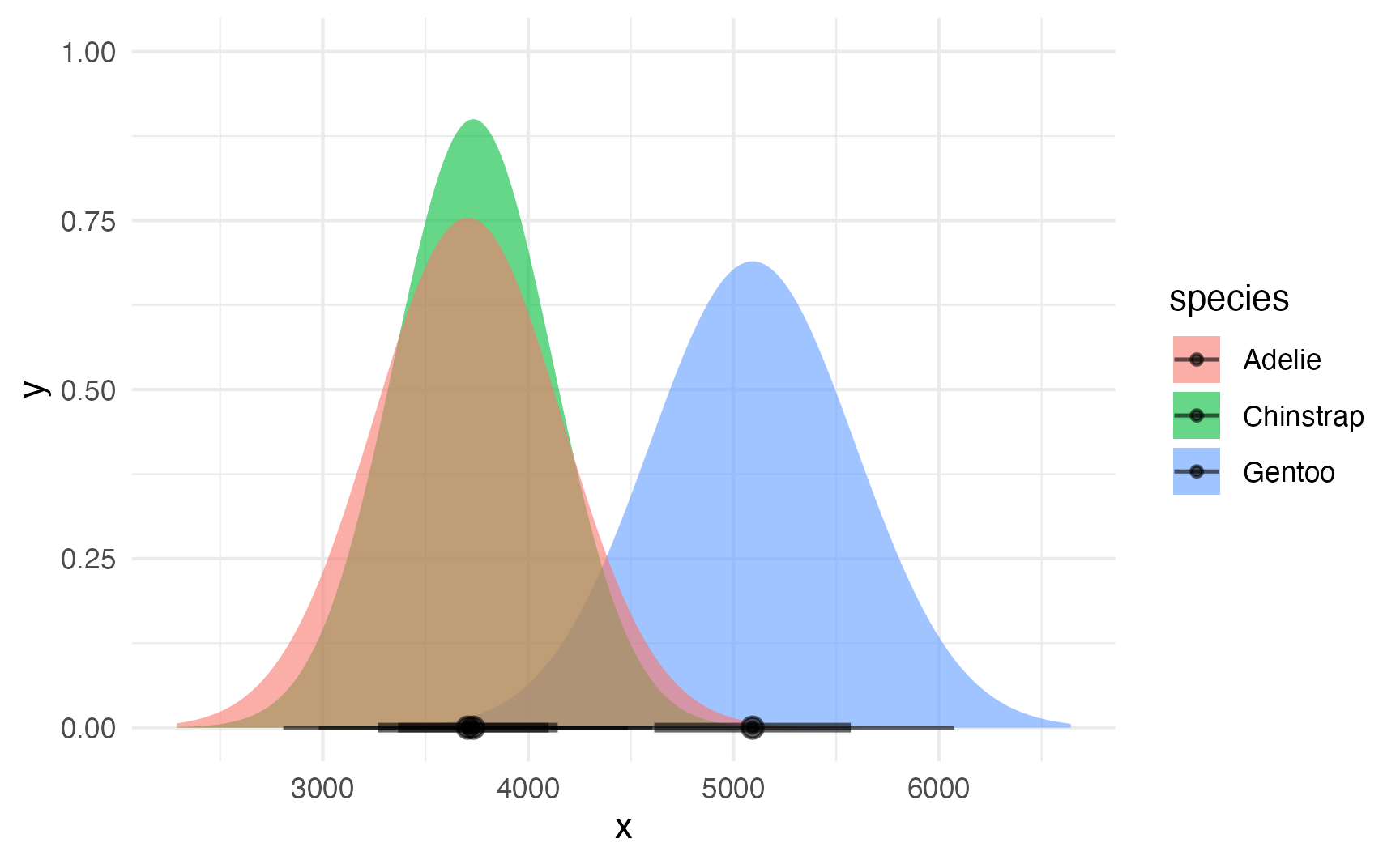
## # ℹ 1 more variable: dist <dist>Plotting this is much easier with the {ggdist} package:
ggplot() +
stat_slabinterval(
data = penguin_parameters_dist,
aes(dist = dist, fill = species, y = 0),
alpha = 0.6
)
We can even combine these estimated density plots with the actual data densities with some minor tweaks to the function arguments (and adjusting the y-axis range):
ggplot(penguins, aes(x = body_mass_g, color = species)) +
stat_density(geom = "line", position = position_identity()) +
stat_slabinterval(
data = penguin_parameters_dist,
aes(dist = dist, fill = species, y = 0),
alpha = 0.4, normalize = "none", inherit.aes = FALSE
) +
coord_cartesian(ylim = c(0, 0.0011))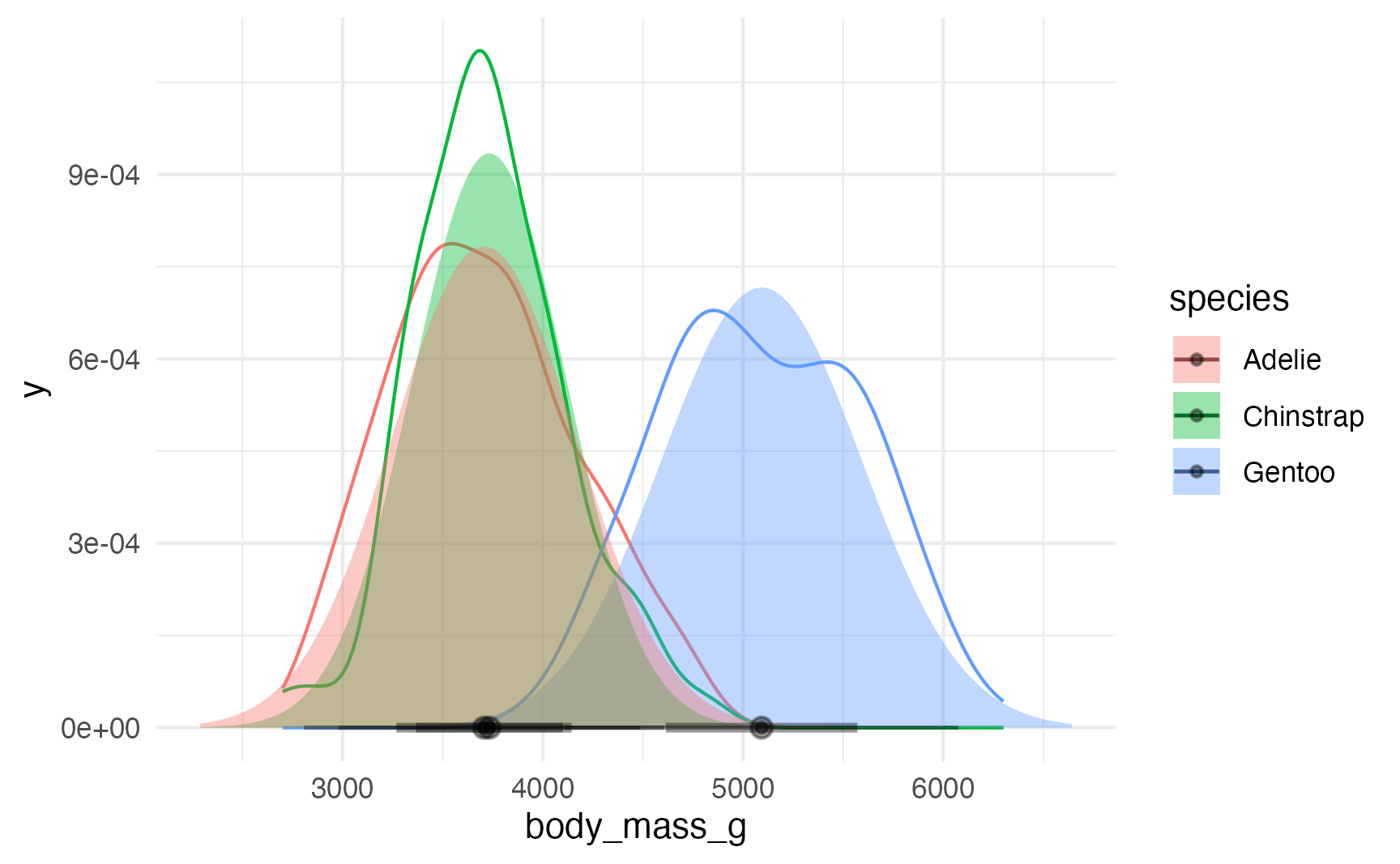
Explicit distributional regression with Bayesianism
It’s neat that we can get the \(\mu\) and \(\sigma\) out of a regular OLS regression model and think about the outcomes as distributions. We can also use continuous predictors to show how the \(\mu\) and \(\sigma\) change across different values of those predictors, but getting that information out of the results of lm() requires a bunch of work.
Fortunately, Bayesian regression (with the magical {brms} package) lets us explicitly model both the \(\mu\) and—optionally—the \(\sigma\).
\[ \begin{aligned} \text{Penguin weight}_i &\sim \mathcal{N}(\mu_i, \sigma_i) \\ \mu_i &= \beta_0 + \beta_{1 \dots 2}\ \text{Species}_i \end{aligned} \]
model_bayes <- brm(
bf(body_mass_g ~ species),
data = penguins,
family = gaussian,
chains = 4, iter = 2000, seed = 1234,
file = "models/model_ols_bayes", refresh = 0
)
model_bayes
## Family: gaussian
## Links: mu = identity; sigma = identity
## Formula: body_mass_g ~ species
## Data: penguins (Number of observations: 333)
## Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
## total post-warmup draws = 4000
##
## Regression Coefficients:
## Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
## Intercept 3705.19 37.72 3631.14 3780.04 1.00 3455 2930
## speciesChinstrap 30.28 67.42 -104.26 158.56 1.00 3506 2919
## speciesGentoo 1387.41 58.25 1273.82 1504.04 1.00 3777 2992
##
## Further Distributional Parameters:
## Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
## sigma 462.16 18.03 428.23 498.85 1.00 3853 3221
##
## Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
## and Tail_ESS are effective sample size measures, and Rhat is the potential
## scale reduction factor on split chains (at convergence, Rhat = 1).model_parameters(model_bayes, verbose = FALSE)
## # Fixed Effects
##
## Parameter | Median | 95% CI | pd | Rhat | ESS
## --------------------------------------------------------------------------
## (Intercept) | 3704.87 | [3631.14, 3780.04] | 100% | 1.000 | 3434.00
## speciesChinstrap | 31.75 | [-104.26, 158.56] | 67.07% | 1.000 | 3485.00
## speciesGentoo | 1387.17 | [1273.82, 1504.04] | 100% | 0.999 | 3774.00
##
## # Sigma
##
## Parameter | Median | 95% CI | pd | Rhat | ESS
## --------------------------------------------------------------
## sigma | 461.66 | [428.23, 498.85] | 100% | 1.001 | 3814.00model_bayes |>
gather_draws(`^b_.*`, regex = TRUE) |>
ggplot(aes(x = .value, fill = .variable)) +
stat_halfeye(normalize = "xy") +
labs(x = "Coefficient value", y = NULL) +
facet_wrap(vars(.variable), scales = "free_x") +
guides(fill = "none") +
theme(axis.text.y = element_blank())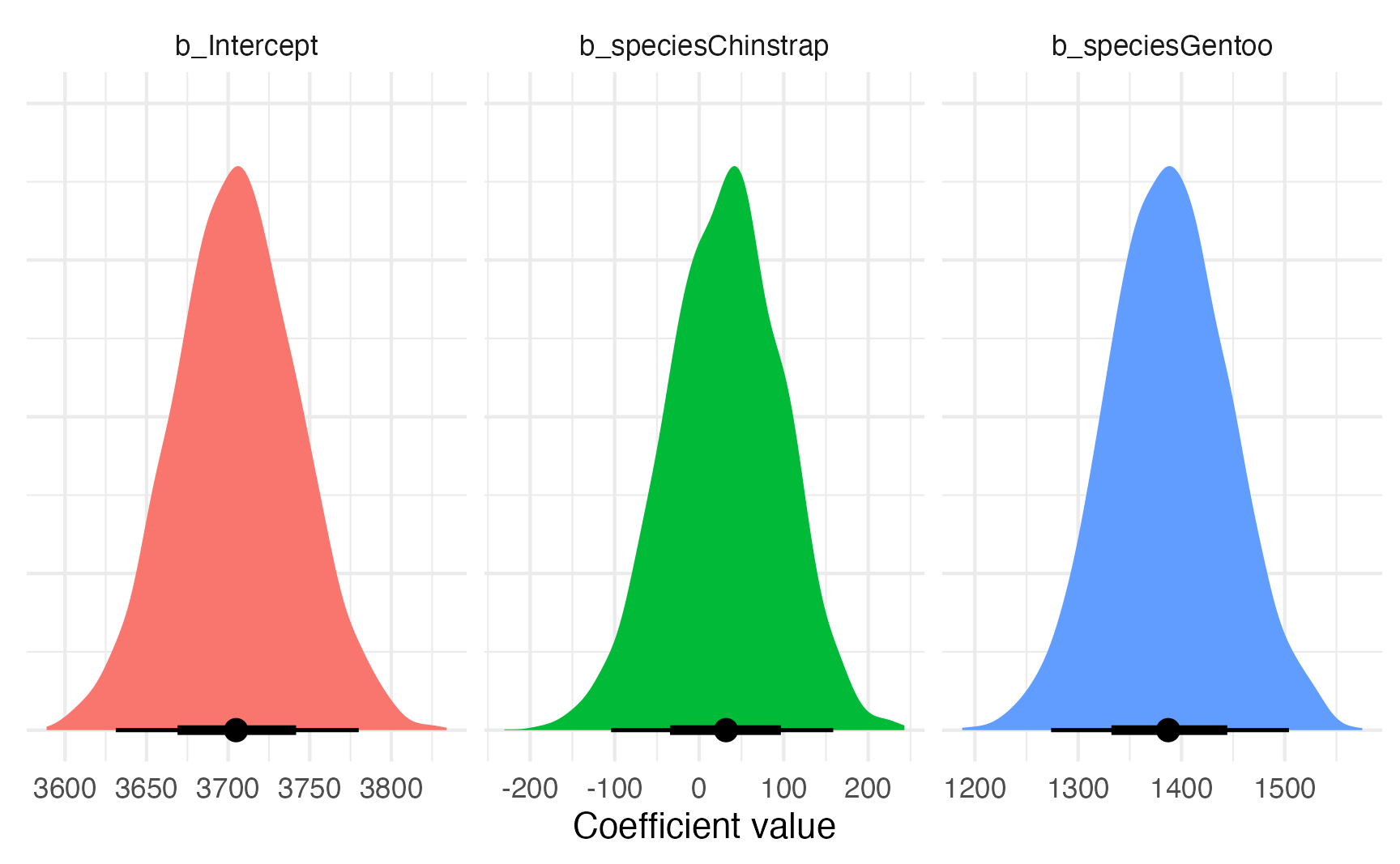
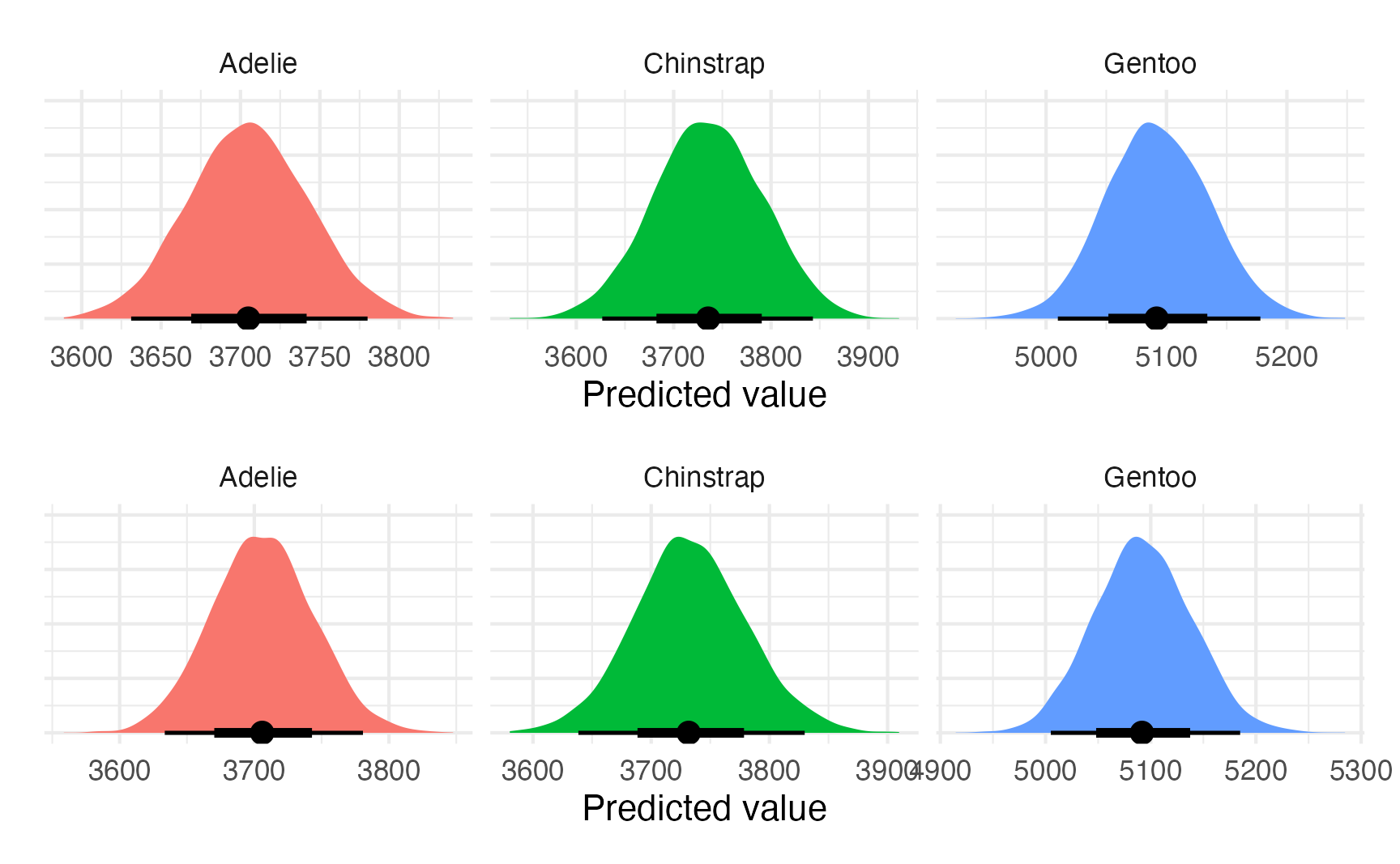
plot_predictions_equal_variances <- model_bayes |>
avg_predictions(variables = "species") |>
posterior_draws() |>
ggplot(aes(x = draw, fill = species)) +
stat_halfeye(normalize = "xy") +
labs(x = "Predicted value", y = NULL) +
facet_wrap(vars(species), scales = "free_x") +
guides(fill = "none") +
theme(axis.text.y = element_blank())
plot_predictions_equal_variances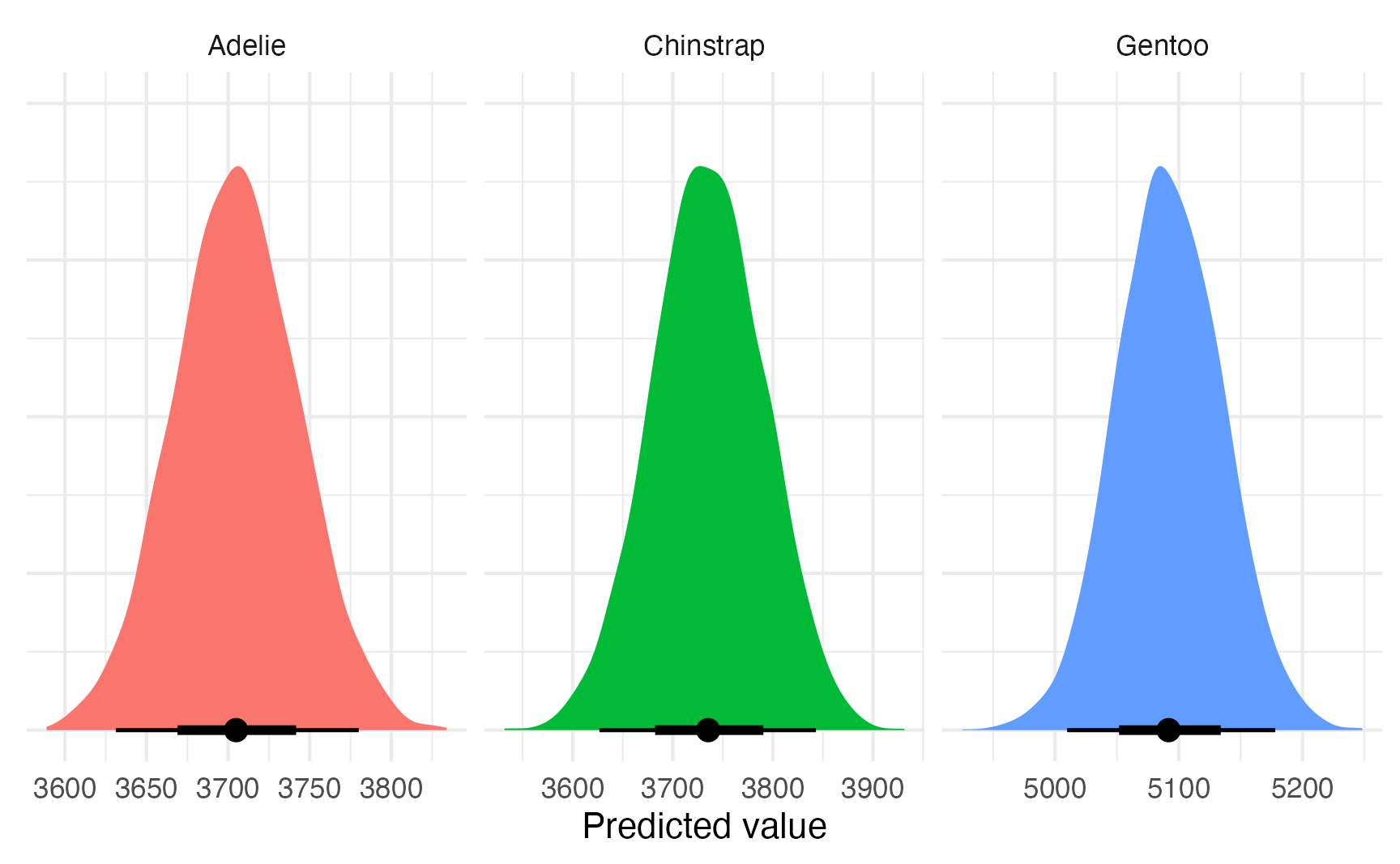
We can also explicitly model the \(\sigma\) part of the outcome, and we can even model it differently from the \(\mu\) part if we have reason to believe the process might be different. For simplicitly, we’ll just use the same explanatory variables for both parts:
\[ \begin{aligned} \text{Penguin weight}_i &\sim \mathcal{N}(\mu_i, \sigma_i) \\ \mu_i &= \beta_0 + \beta_{1 \dots 2}\ \text{Species}_i \\ \log(\sigma_i) &= \gamma_0 + \gamma_{1 \dots 2}\ \text{Species}_i \end{aligned} \]
model_bayes_sigma <- brm(
bf(
body_mass_g ~ species,
sigma ~ 0 + species
),
data = penguins,
family = gaussian,
chains = 4, iter = 2000, seed = 1234,
file = "models/model_ols_sigma_bayes", refresh = 0
)
model_bayes_sigma
## Family: gaussian
## Links: mu = identity; sigma = log
## Formula: body_mass_g ~ species
## sigma ~ 0 + species
## Data: penguins (Number of observations: 333)
## Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
## total post-warmup draws = 4000
##
## Regression Coefficients:
## Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
## Intercept 3706.19 37.83 3633.52 3780.70 1.00 4935
## speciesChinstrap 26.77 60.10 -89.23 147.82 1.00 4926
## speciesGentoo 1386.56 59.41 1274.78 1501.10 1.00 4735
## sigma_speciesAdelie 6.13 0.06 6.02 6.25 1.00 5642
## sigma_speciesChinstrap 5.96 0.09 5.79 6.14 1.00 4898
## sigma_speciesGentoo 6.22 0.06 6.10 6.35 1.00 5747
## Tail_ESS
## Intercept 3182
## speciesChinstrap 3354
## speciesGentoo 3154
## sigma_speciesAdelie 3212
## sigma_speciesChinstrap 3054
## sigma_speciesGentoo 3546
##
## Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
## and Tail_ESS are effective sample size measures, and Rhat is the potential
## scale reduction factor on split chains (at convergence, Rhat = 1).model_parameters(model_bayes_sigma, verbose = FALSE)
## # Fixed Effects
##
## Parameter | Median | 95% CI | pd | Rhat | ESS
## --------------------------------------------------------------------------------
## (Intercept) | 3705.86 | [3633.52, 3780.70] | 100% | 1.001 | 4903.00
## speciesChinstrap | 25.79 | [ -89.23, 147.82] | 66.77% | 1.000 | 4867.00
## speciesGentoo | 1385.96 | [1274.78, 1501.10] | 100% | 1.000 | 4731.00
## sigma_speciesAdelie | 6.13 | [ 6.02, 6.25] | 100% | 1.000 | 5667.00
## sigma_speciesChinstrap | 5.96 | [ 5.79, 6.14] | 100% | 1.000 | 4805.00
## sigma_speciesGentoo | 6.22 | [ 6.10, 6.35] | 100% | 1.000 | 5670.00Now we have model parameters for the regular coefficients and the sigmas (prefixed with sigma_). For whatever reason, Stan works with these on the log scale, which is why they all look really tiny. We can unlog them by exponentiating them:
model_parameters(
model_bayes_sigma,
keep = "sigma_",
exponentiate = TRUE,
verbose = FALSE
)
## # Fixed Effects
##
## Parameter | Median | 95% CI | pd | Rhat | ESS
## ---------------------------------------------------------------------------
## sigma_speciesAdelie | 460.06 | [412.32, 517.71] | 100% | 1.000 | 5667.00
## sigma_speciesChinstrap | 385.97 | [326.90, 464.30] | 100% | 1.000 | 4805.00
## sigma_speciesGentoo | 502.30 | [444.81, 573.58] | 100% | 1.000 | 5670.00They’re the same as what we found earlier by calculating the standard deviation of the residuals!
penguin_parameters
## # A tibble: 3 × 3
## species mu sigma
## <fct> <dbl> <dbl>
## 1 Adelie 3706. 459.
## 2 Chinstrap 3733. 384.
## 3 Gentoo 5092. 501.plot_predictions_different_variances <- model_bayes_sigma |>
avg_predictions(variables = "species") |>
posterior_draws() |>
ggplot(aes(x = draw, fill = species)) +
stat_halfeye(normalize = "xy") +
labs(x = "Predicted value", y = NULL) +
facet_wrap(vars(species), scales = "free_x") +
guides(fill = "none") +
theme(axis.text.y = element_blank())
plot_predictions_different_variances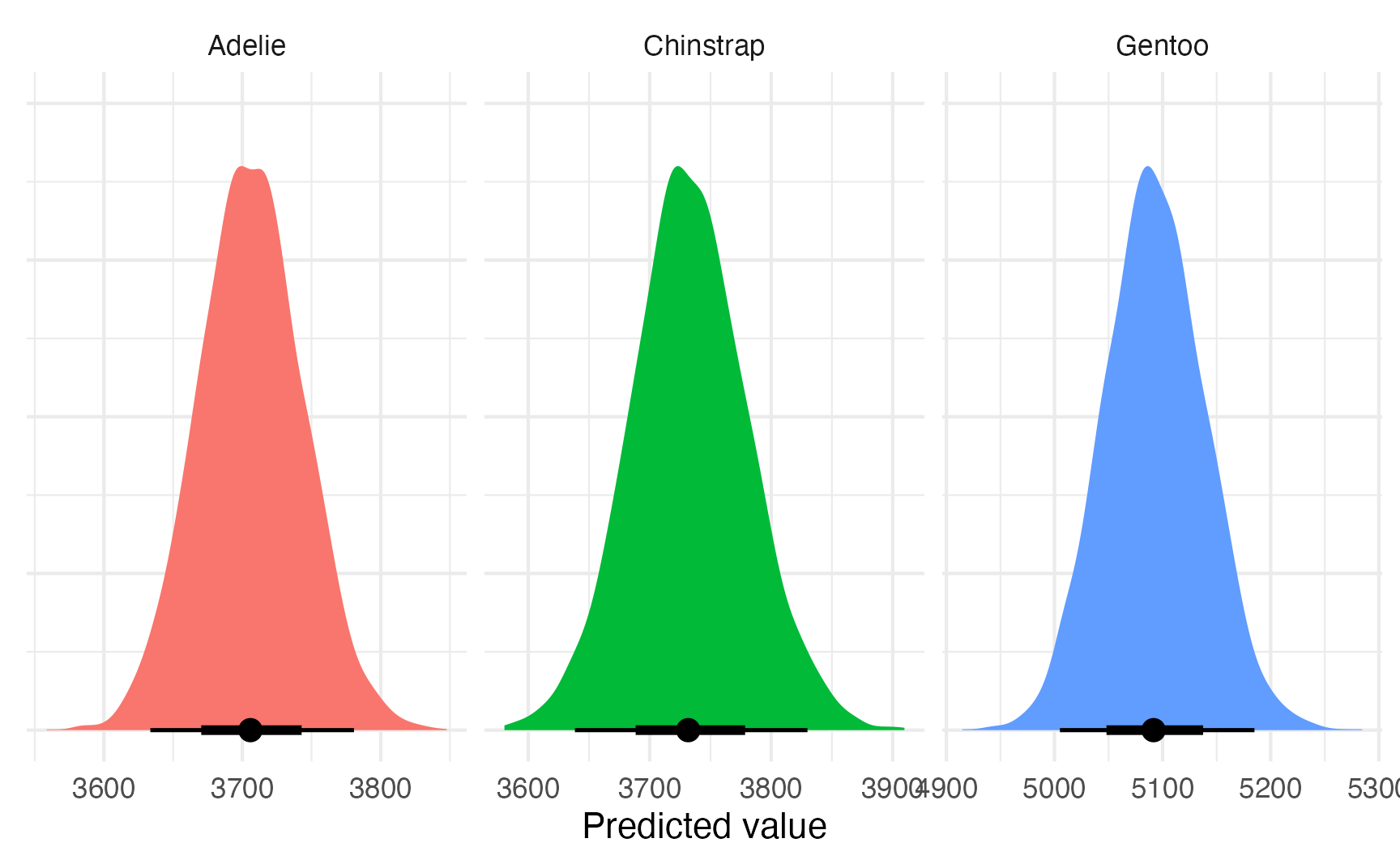
(plot_predictions_equal_variances + labs("Same sigma")) /
(plot_predictions_different_variances + labs("Different sigmas"))